The use of CRISPR (clustered regularly interspaced short palindromic repeats) and associated Cas enzymes for genome editing has been a major technological breakthrough, making genome modification in cells or organisms faster, more efficient, and more robust than previous genome editing methods such as zinc finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs).
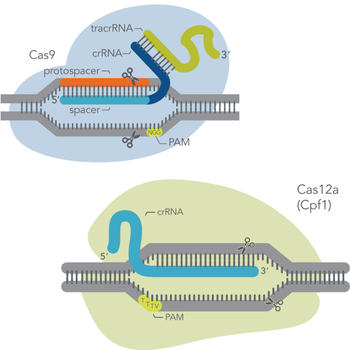
The most commonly used system combines a guide RNA (gRNA) with the Cas9 enzyme from Streptococcus pyogenes to achieve gene disruption or modification via knock-in or knock-out strategies. Other approaches exist, making use of e.g. modified Cas9 variants like nickases, dead variants, or Cas9 fusions to additional DNA modifying enzymes. In addition alternative Cas enzymes are available, such as Cas12a (also termed Cpf1) from Acidaminococcus sp. BV3L6 or Lachnospiraceae bacterium ND2006, which allow targeting of gene locations inaccessible to the Cas9 system.

Components and mechanism of action
At the most basic level, CRISPR genome editing requires two components: a Cas enzyme (an endonuclease) and a guide RNA. These components associate to form a ribonucleoprotein (RNP) complex. Then, the guide RNA can base pair with a protospacer (i.e., the target genomic sequence of about 20 bases in length), which is adjacent to a genomic sequence called a protospacer-adjacent motif (PAM). The PAM is only a few bases long, and its sequence depends on the Cas enzyme used. Once the RNP binds to the target DNA (protospacer), the Cas enzyme makes a precise cut in the target. The type of cut – blunt-ended or staggered double-strand break (DSB) – depends on the Cas enzyme used.
Terminology can be confusing. Here, as in the context of most laboratory genome editing, the protospacer is the target DNA sequence. The spacer is the section of the guide RNA which base pairs with the target. The picture on the right depicts the different components of the CRISPR-Cas9 system.

Types of genome edits: knock-ins vs knock-outs
Once the Cas enzyme complexed to a guide RNA is delivered into the cells, it is directed by the gRNA to a user-specified cut site in the genome. There it introduces a double-strand break. Cells will try to repair DSBs, because unrepaired breaks can lead to cell death due to loss of genome stability. Two outcomes are possible:
Non-homologous end joining (NHEJ)
Non-homologous end-joining is the best known and probably the most common repair mechanism that makes changes (mutations) in the DNA [1]. In addition, other pathways, such as alternative end-joining [2], single-strand annealing [3], and other cellular mechanisms, can also result in DNA repair with mutations. As the name NHEJ implies, the NHEJ pathway joins DSB ends without the need for a homologous template. During the processes of ligating the cut ends of the DNA, NHEJ can often introduce small indels at the break point (see figure on the right). Often an indel causes a frameshift mutation, which can disrupt a gene’s expression and function, which is very useful when you want to knock out a target gene.
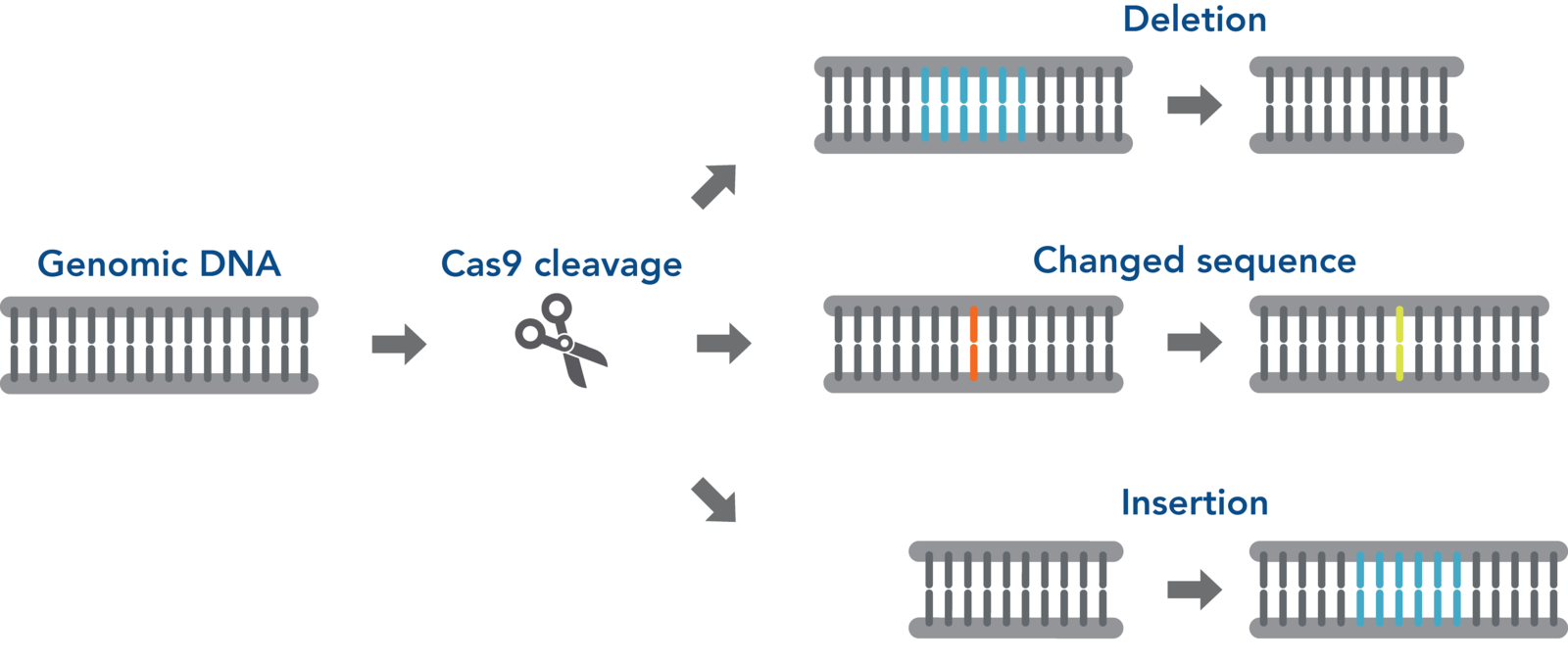
Homology-directed repair (HDR)
In addition to mutagenic pathways for DSB repair, cells may use HDR, a non-mutagenic mechanism. HDR is a broad category that includes many distinct pathways leading to recombination [4–6]. Most of the HDR pathways lead to perfect copying of a donor strand into the genomic sequence being repaired. HDR seals a DSB in an error-free fashion, but not necessarily with the original sequence. Gene knock-in may be desired by researchers aiming to understand gene function or model disease-causing mutations. Generally speaking, knock-in experiments involve HDR for effectively and precisely introducing an exogenous DNA fragment. HDR relies on a DNA template (donor) with sections homologous to the DNA that will be repaired or changed. When there is a DSB due to CRISPR cutting of DNA, a donor must have ends that are homologous with the DNA near the ends of the DSB. The new sequence between the two homologous ends will be incorporated into the genome.

The HDR donor template must be delivered along with the guide RNA:Cas nuclease ribonucleoprotein complex. If no donor DNA strand is supplied, NHEJ will be the predominant or only mechanism of DSB repair. However, even when a donor DNA strand is supplied, the error-free HDR pathway is inefficient compared to NHEJ. HDR rates vary widely between cell types but usually are much lower than the rates of NHEJ. LubioScience's supplier Integrated DNA Technologies (IDT) has developed several reagents and protocols that greatly enhance error-free HDR rates.
CRISPR applications beyond simple editing
CRISPR screening
CRISPR screening identifies a small number of genes (out of the whole genome) involved in a specific physiological effect. Most CRISPR screening is done in cell culture, although some methods have been devised for use in animal models. In CRISPR screening, scientists usually knock out every gene in the genome that could be important, knocking out only one gene per cell. During this process, some cells die, but others survive and become the predominant cell types. Then, the scientists do next generation sequencing (NGS) on the surviving cells to find out which sequences are still present.
CRISPR without double-strand DNA breaks
Gene silencing
Almost immediately after the discovery of CRISPR genome editing, some researchers produced a mutated Cas9 that could not cut DNA. This catalytically inactive enzyme, dCas9 (dead Cas9), could still be targeted to a specific genomic site. Interestingly, simply by binding to a target site, dCas9 was able to inactivate gene transcription at that site by preventing binding of the cellular transcription machinery to the gene [7]. The gene is effectively silenced until the cell naturally eliminates the dCas9 enzyme. This approach, whereby CRISPR is used to temporarily silence a gene without cutting the DNA, was named CRISPR interference (CRISPRi).
Gene activation
CRISPRa is a category of methods that use the same dead Cas9 mutant as CRISPRi. However, in CRISPRa, the RNP is used to carry transcriptional activators, which overcome the transcription-blocking effect caused by dCas9, and turn on transcription at target promoter regions within the target gene. Some CRISPRa systems use activating proteins connected to the gRNA itself rather than to the Cas enzyme [8]. Like CRISPRi, CRISPRa methods have been extensively developed so that transcriptional activity can be tightly modulated, such as in response to light [9]. Also, both CRISPRi and CRISPRa can be used to perform powerful genome-wide screens.
Other non-editing functions with dCas9-containing RNP
Because dCas9 can target specific genomic sites without cutting the target, dCas9 is useful when fused to other proteins. As part of an RNP, dCas9 can transport activators, inhibitors, fluorescent proteins, and epigenomic modifier enzymes to specific sites in the genome to achieve numerous desired effects at those sites.
Nuclear organization and epigenetic modification
Fluorescently labelled CRISPR components can help study nuclear organization, enabling easy visualization of target genes within the nucleus. Numerous systems using modifications of CRISPR components have been designed for this purpose [10]. The CRISPR technology can also be used to direct enzymes to particular sites in the genome to regulate epigenetic modifications [9].
What do I need for my genome editing experiment?
To begin a CRISPR genome editing experiment, you will need a guide RNA, a Cas nuclease, and an appropriate delivery system. One key decision is selecting the CRISPR system best suited to your application. If your experiment involves HDR, a donor DNA template is also required. Additionally, proper experimental design includes the use of appropriate controls, and in some cases, suitable enhancers to improve efficiency. Finally, analyzing gene editing outcomes necessitates the use of reliable and suitable methods.
LubioScience provides a wide range of high-quality products from trusted suppliers to support the success of your genome editing project. Use the navigation below to explore our portfolio of CRISPR genome editing products tailored to your specific needs.
Selecting a CRISPR-Cas System
Link for Selecting a CRISPR-Cas System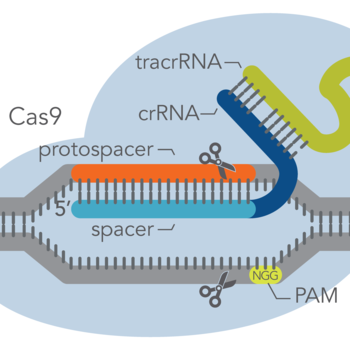
The CRISPR-Cas9 system
Link for Gene Editing with the CRISPR-Cas9 SystemThe CRISPR Starter Kit
Link for The CRISPR Starter Kit
The CRISPR-Cas12a system
Link for Gene Editing with the CRISPR-Cas12a System
Knock-Ins via HDR
Link for Knock-Ins via HDR
RNP Complex Delivery
Link for RNP Complex Delivery
Analysis of gene edits
Link for Analysis of CRISPR-Cas Gene Edits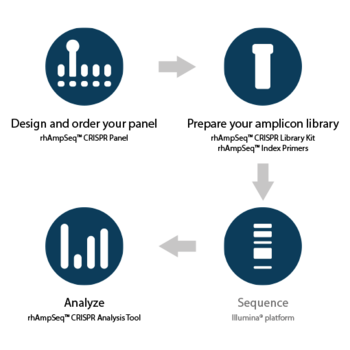
rhAmpSeq™ CRISPR Analysis System
Link for rhAmpSeq™ CRISPR Analysis System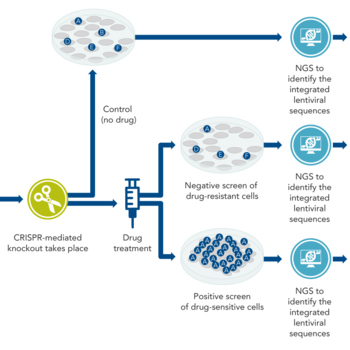
CRISPR-based screens
Link for CRISPR-Based Genome-Wide Functional ScreeningFurther Reading and Links
Link for Further Reading and LinksCustom guide RNAs
We offer CRISPR custom guide RNAs which are suitable for common as well as specialized research applications. They are ideal for prime editing (pegRNA) projects, CRISPR-Cas13 applications, and most alternative CRISPR-Cas systems. Benefit from a range of modification options and multi-scale delivery in tubes or plates.
New in the field of CRISPR-Cas genome editing?
Get a discount on your first order!
CRISPR cGMP gRNA manufacturing service and GMP CRISPR nucleases
Are you looking to accelerate your CRISPR genome editing program from discovery to clinical trials? IDT's Engineering Run and full cGMP guide RNA manufacturing services offer a streamlined and regulatory-compliant solution. Backed by comprehensive documentation, their guide RNAs simplify regulatory filings, offering a straightforward path to clinical success.
Does your project require high-quality cGMP nucleases? IDT offers SpCas9 Nuclease and SpyFi® Cas9 Nuclease – available for immediate delivery at cGMP quality.
IDT's GMP services and products are not provided via LubioScience. If you are interested, please contact IDT directly.
Haven't found what you are looking for?
References
- Chang HHY, Pannunzio NR, et al. (2017) Non-homologous DNA end joining and alternative pathways to doublestrand break repair. Nat Rev Mol Cell Biol 18(8):495–506.
- Sallmyr A and Tomkinson AE (2018) Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem 293(27):10536–10546.
- Bhargava R, Onyango DO, et al. (2016) Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32(9):566–575.
- Chen J-M, Férec C, et al. (2010) Gene conversion in human genetic disease. Genes 1(3):550–563.
- Verma P and Greenberg RA (2016) Noncanonical views of homology-directed DNA repair. Genes Dev 30(10):1138–1154.
- Savic N, Ringnalda FC, et al. (2018) Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 7: e33761.
- Qi LS, Larson MH, et al. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173–1183.
- Konermann S, Brigham MD, et al. (2014) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583.
- Polstein LR and Gersbach CA (2015) A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol 11:198.
- Hong Y, Lu G, et al. (2018) Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging. Genome Biol 19(1):39.
For research use only. Not for use in diagnostic procedures. Unless otherwise agreed to in writing, IDT does not intend for these products to be used in clinical applications and does not warrant their fitness or suitability for any clinical diagnostic use. Purchaser is solely responsible for all decisions regarding the use of these products and any associated regulatory or legal obligations.