Introduction
Since its initial release in 2000, the human reference genome has covered only the euchromatic fraction of the genome, leaving heterochromatic regions - representing 8% of the human genome - unfinished. Now, the Telomere-to-Telomere (T2T) Consortium presents a complete 3.055 billion–base pair sequence of a human genome, that includes gapless assemblies for all chromosomes except Y and introduces nearly 200 million base pairs of additional sequence, including 99 predicted protein coding genes.
The challenge
The current human reference genome was released by the Genome Reference Consortium (GRC) in 2013. This reference traces its origin to the publicly funded Human Genome Project and has been continually improved over the past two decades. Unlike most modern sequencing projects based on “shotgun” sequence assembly the GRC assembly was constructed from sequenced bacterial artificial chromosomes (BACs) that were ordered and oriented along the human genome by means of radiation hybrid, genetic linkage, and fingerprint maps. However, limitations of BAC cloning led to an underrepresentation of repetitive sequences, and the opportunistic assembly of BACs derived from multiple individuals resulted in a mosaic of haplotypes. As a result, several GRC assembly gaps are unsolvable because of incompatible structural polymorphisms on their flanks, and many other repetitive and polymorphic regions were left unfinished or incorrectly assembled.
As a result, the GRCh38 reference assembly contains 151 mega–base pairs (Mbp) of unknown sequence distributed throughout the genome,. Some of the largest reference gaps include human satellite (HSat) repeat arrays and the short arms of all five acrocentric chromosomes, which are represented in GRCh38 as multimegabase stretches of unknown bases. In addition to these apparent gaps, other regions of GRCh38 are artificial or are otherwise incorrect. When compared with other human genomes, GRCh38 also shows a genome-wide deletion bias that is indicative of incomplete assembly. Despite finishing efforts from both the Human Genome Project and GRC, there was limited progress toward closing the remaining gaps in the years that followed.
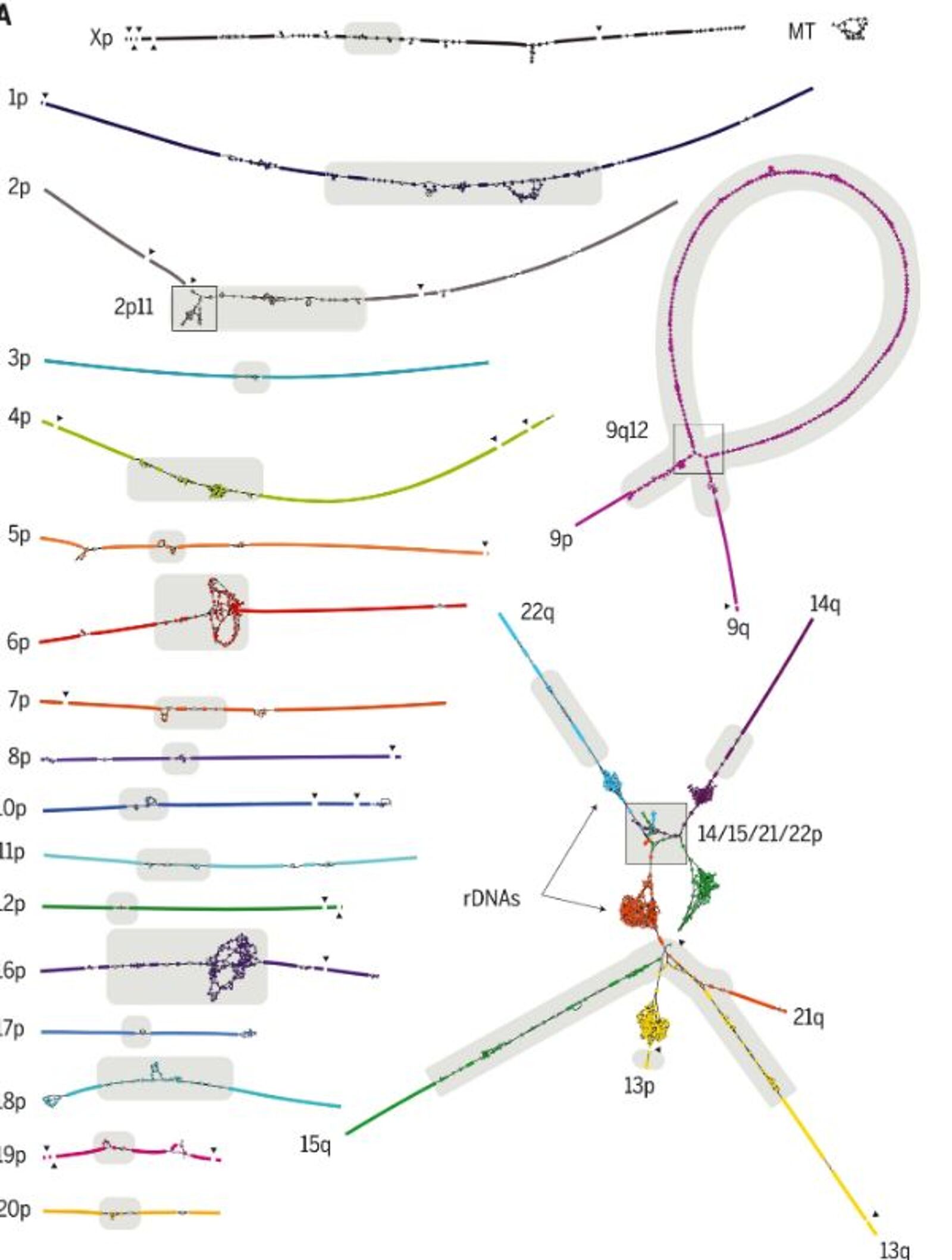
Key results
The Telomere-to-Telomere Consortium used multiple long-read shotgun sequencing techniques to overcome the limitations of BAC-based assembly and to bypasse the challenges of structural polymorphism between genomes. PacBio’s multikilobase, single-molecule reads proved capable of resolving complex structural variation and gaps in GRCh38 whereas Oxford Nanopore’s >100-kbp “ultralong” reads enabled complete assemblies of a human centromere and, later, an entire chromosome. However, the high error rate (>5%) of these technologies posed challenges for the assembly of long, near-identical repeat arrays. The use of PacBio’s most recent “HiFi” circular consensus sequencing offered a solution to this dilemma, allowing for 20-kbp read lengths with a reduced error rate of 0.1%. Whereas ultralong reads proved to be useful for spanning repeats, HiFi reads excelled at differentiating subtly diverged repeat copies or haplotypes.
To finish the last remaining regions of the genome, the consortium leveraged the complementary aspects of PacBio HiFi and Oxford Nanopore ultralong-read sequencing for final genome assembly. The resulting T2T-CHM13 reference assembly removes a 20-year-old barrier that has hidden 8% of the genome from sequence-based analysis, including all centromeric regions and the entire short arms of five human chromosomes.
Read the original publication
Nurk et al. "The complete sequence of a human genome" Science (2022) 376:6588, p 44-53 DOI: 10.1126/science.abj6987
Check out NGS library prep kits from IDT
IDT offers a wide range of library prep kits, covering all types of NGS applications: