New SOD1-transgenic mouse model validates safety and efficacy of CRISPR Cas9 system

Takeaway: CRISPR genome editing has proven beneficial to our understanding of many diseases, including Amyotrophic Lateral Sclerosis (ALS). Yet, efficacy and long-term safety have remained in question. A recent study builds on prior learnings to secure CRISPR’s staying power as a potential method for future ALS therapeutic endeavors.
Do you remember the Ice Bucket Challenge of 2014? Considering the overwhelming popularity of this challenge, how could you not? Millions of people world-wide answered the seemingly playful ultimatum by choosing to share a video clip of their reaction to ice water being dumped over their head to help raise money for the Amyotrophic Lateral Sclerosis Association. While the motivation behind this outpouring generosity would later be debated, few could argue against whether the campaign succeeded.
To date, the ALS Ice Bucket challenge remains one of the most successful social media-driven awareness and fundraising initiatives of our time, enabling the Association to funnel $111 million toward critical programs, resources, and advancements, including research that paved the way for groundbreaking discoveries years later.
What is ALS?
The National Institute of Neurological Disorders and Stroke (NINDS), defines ALS, also referred to as Lou Gehrig’s disease, as a rare neurological disease that degenerates motor neurons in the central nervous system that are responsible for muscle control. While often originating in the hands, symptoms worsen over time and spread from the brain through the spinal cord to other parts of the body, causing the patient to lose the ability to walk, talk, and chew. Without a cure or a treatment to slow or reverse the disease’s course, prognosis is grim. ALS eventually leads to respiratory failure and death, typically within three years from the start of symptoms.
The root cause of ALS is a mystery. The NINDS estimates up to 90% of cases occur without a trace of family history or underlying genetic abnormality. The remaining 10%, however, inherit the disease through a mutated gene. Understanding how and why this happens for the smaller subset of the patient population may hold the key to unlocking new therapeutics for all ALS patients, regardless of disease origin.
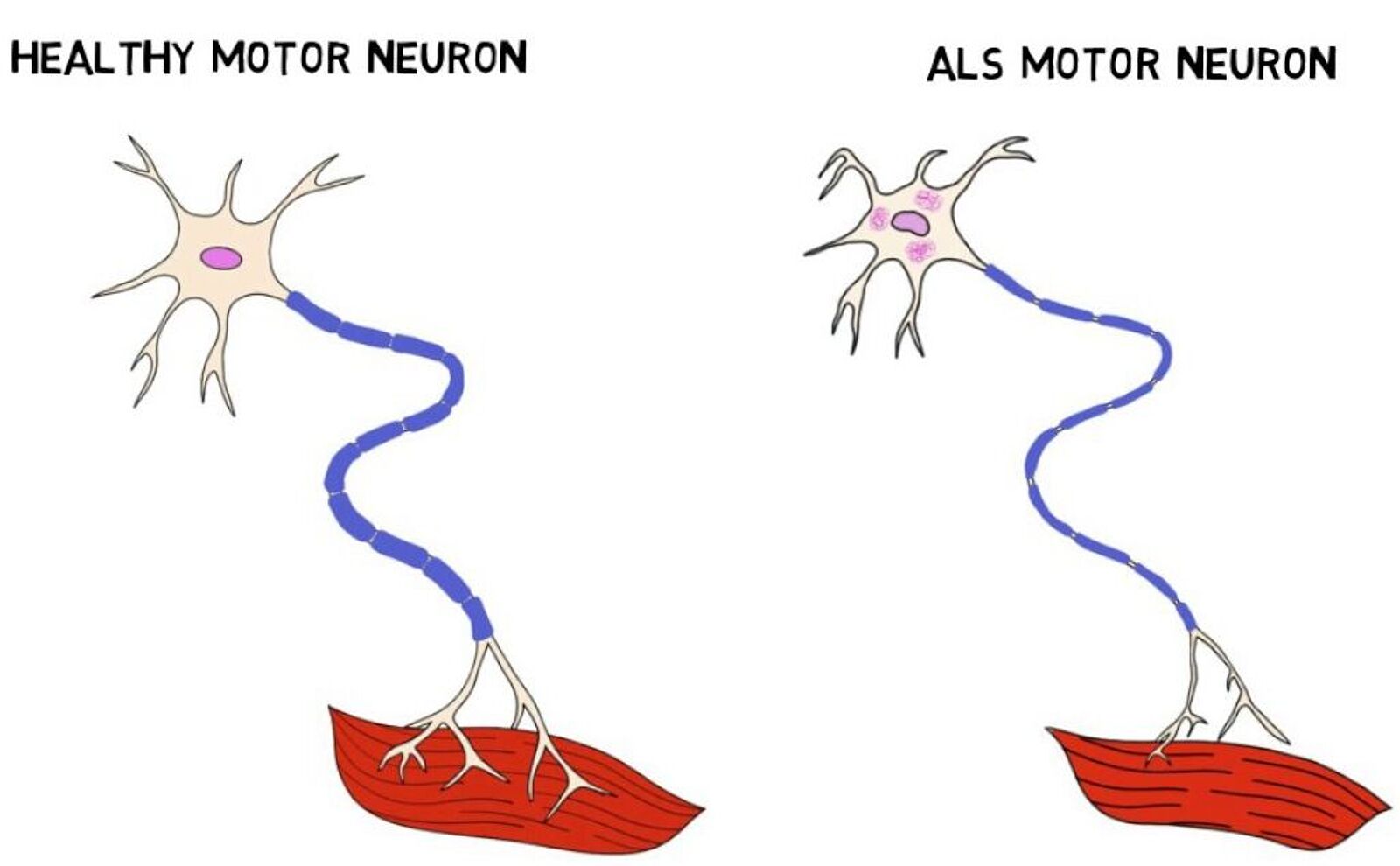
Animal ALS models enable researchers to target SOD1-gene with CRISPR-Cas9
In 1993, scientists supported by the NINDS completed a study on familial genes associated with ALS, indicating the SOD1-amyotrophic lateral sclerosis gene is present in up to 20% of familial ALS cases in North America—giving researchers an important target. Considering this mutation’s history of causing severe disease progression, researchers faced an urgent need to develop new therapeutics that would slow the inevitable.
Since then, additional genome editing studies have proven valuable to expanding our understanding of ALS gene expression and disease outcome. In recent years, the CRISPR-Cas9 system was used to create transgenic mice models with a surplus of the SOD1-gene. Given this model’s pattern of highly predictable disease representation, based in part by the likeness of the mouse’s nervous system to that of humans, this model continues to be the most used.
While aforementioned studies positioned Cas9 as a powerful tool that notably broadened our knowledge of ALS, before the SOD1-gene animal model could be applied to humans, several key challenges remained:
- Low editing efficiency and a limited number of edited cells
- With some instances of off-target effects taking years to present, the short post-edit review windows (weeks and months) result in inconclusive data to support safety over a long period of time
- Onset of tumors associated with lymphoma, sarcoma, or carcinoma caused by unintended off-target effects
- Impact and underlying cause of unintended large (ranging from several hundred to thousand) deletion of DNA at target sites
To solidify future in vitro and in vivo treatment options, scientists would need to address these concerns and validate the potential for long-term safety and efficacy.
CRISRP-Cas9 advances to next step, the successful prevention of ALS-like pathology by CRISPR Cas9-mediated transgene editing
Putting CRISPR-Cas9 technology to work once again, a team led by Dr. Han-Xiang Deng at The Ken and Ruth Davee Department of Neurology at the Feinberg School of Medicine within Northwestern University set out to investigate whether the CRISPR genome editing approach for ALS could be amplified for maximum benefit while also maintaining safety and efficacy that would withstand the test of time.
Recently published in Nature, Efficacy and long-term safety of CRISPR-Cas9 genome editing in the SOD1-linked mouse models of ALS outlines the team’s work using long-range PCR, cloning, Sanger-sequencing approaches, design tools like IDT’s CRISPR-Cas9 RNA Design Checker, and the SOD1-G93A model as a blueprint to carefully design two distinct models that would apply learnings from prior studies to edit the gene using single guide RNA (sgRNA) and CRISPR-Cas9 in early embryonic stages. This in vivo approach would allow the team to study gRNA and CRISPR-Cas9 expression throughout the lifespan of the mice. The anticipated result: maximum editing efficiency and minimum occurrence of adverse effects to validate the long-term efficacy and safety of this approach.
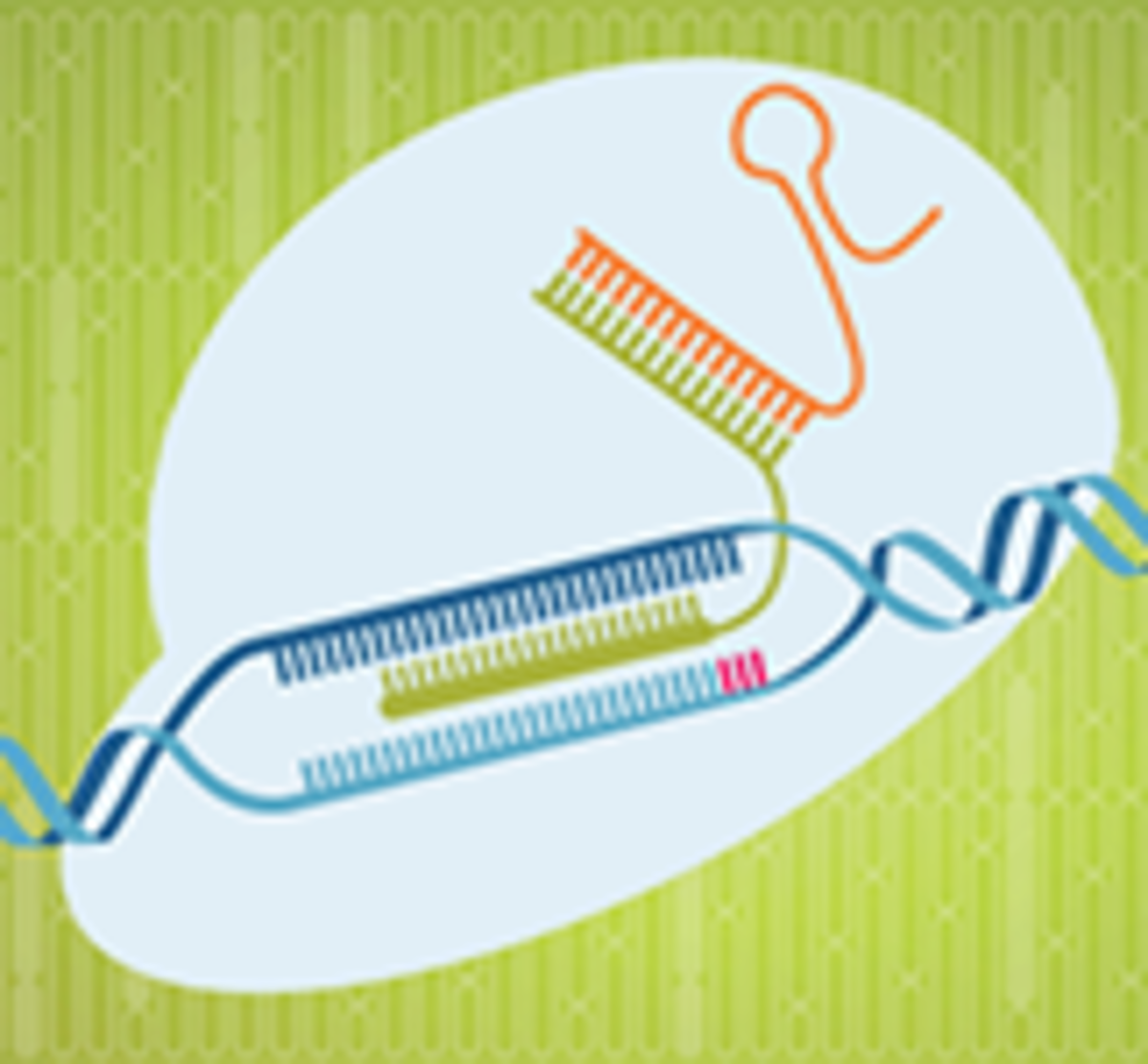
While the team did observe off-target events, these occurrences appeared to be highly predictable, allowing the team at Northwestern to design adjustments to their models to combat these issues. An example of the off-target events include mismatches in the gRNA near the targeted site, which the team believed could be less tolerated in vivo vs. in vitro, due to the higher concentration found in the in vitro system. Another was deletions occurring in the Alu sequences, which have no know functions and are not associated with disease.
Upon conclusion, data confirmed the CRISPR-Cas9 gene-editing approach to be highly successful and completely prevent any clinical or pathological signs of ALS. Off-target effects were predictable, targetable, and low-risk. Combined, these data suggest long-term safety and efficacy of using CRISPR-Cas9 to edit the SOD1-gene.
The future for ALS
Successfully curing mice of the ALS gene, while validating long-term safety and efficacy of the CRISPR-Cas9 system, is a big step in the right direction. This important work adds another layer to a growing foundation for which future scientists can continue to target and explore how CRISPR technologies may be deployed in similar way in humans. In the meantime, the ALS Association continues to monitor and invest in new approaches to identify new therapeutics and treatment plans for this devastating disease. And the ice bucket challenge? Yes, it’s still a thing.
To learn more about how IDT can support your critical CRISPR genome editing research, visit the IDT CRISPR hub today.