This blog is the first part of a two-part series on spatial proteomics. Read part 2 here.
Proteomics
The term proteomics describes the study of the structure and function of all proteins produced by a cell, tissue or organism under specific conditions. Unlike the genome, which remains defined for each organism, the proteome is subject to change depending on the tissue and cell type and cellular activities. Although the gene sequence dictates a protein sequence, proteins can undergo alternative splicing — where exons (the genomic region that ends up within the mRNA molecule) are assembled in different combinations during mRNA splicing (when introns are removed, and exons are put together). Additional changes can occur in the form of post-translational modifications (PTMs). Therefore, the number of proteins far exceeds the number of genes present.
The term proteomics was only coined in the mid-1990s(1), but the field of proteomics had existed for many decades before. The first proteomics studies began in the mid-1900s using two-dimensional gel electrophoresis (2-DE) for studying proteins from E. coli(2). 2-DE allows proteins to be separated according to their isoelectric point (pI) — the pH at which the protein has no overall charge — and their molecular weight. 2-DE allows further fractionation of proteins compared to 1D gel electrophoresis. Due to its superior ability to resolve proteins, 2-DE can separate proteins containing in vivo modifications and missense mutations that lead to differences in protein charge. Other techniques are used for proteomics, including yeast-two analysis and protein microarrays.
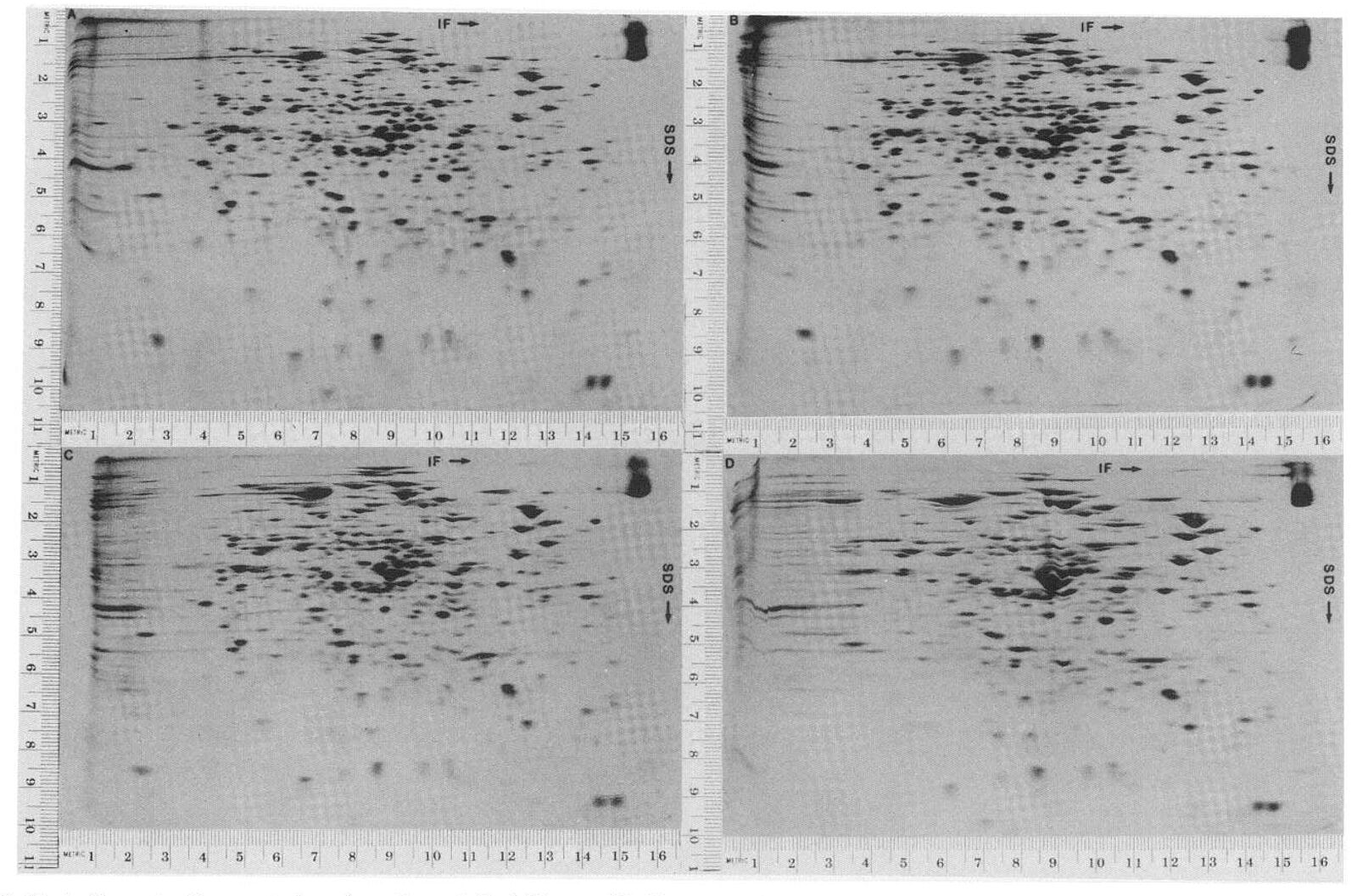
High-throughput proteomic analysis using mass spectrometry
Mass spectrometry (MS) has been instrumental in high-throughput proteomic analysis. The first MS instrument was invented in 1912 and was used for isotopic analysis. It was not until 1996 that MS was applied to protein analysis(3). MS involves digesting proteins into peptides, performing separation techniques (either with gels or chromatography), and fragmenting and ionizing samples to measure the mass/charge (m/z) ratio of gas-phase ions. There are two main ways to generate ions:
- Matrix-assisted laser desorption/ionization (MALDI) — which produces ions from a matrix that absorbs the laser energy,
- Electrospray — which produces ions when a high voltage is applied to a liquid. The ionization source is coupled with mass analyzers. Examples of analyzers include Time-of-flight (TOF), quadrupole and quadrupole ion trap. Multiple rounds of fragmentation and mass analysis can be used to elucidate peptide sequencing, known as tandem MS.
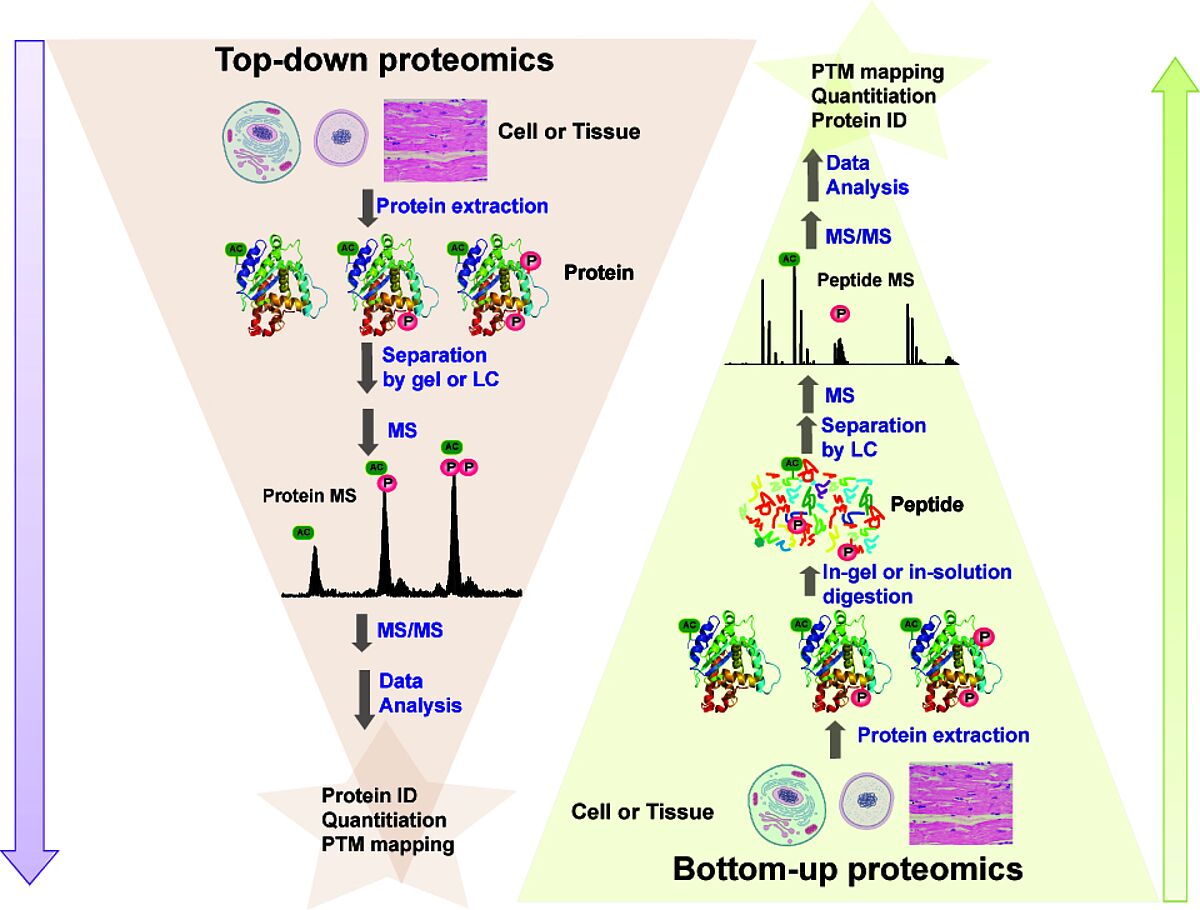
Proteins can be identified by the resulting data — a mass spectrum. Identification is performed by comparing experimental data with databases containing theoretical spectra from a protein sequence database. A probability score can be used to indicate how much confidence can be placed in the assignment. Potential issues with protein databases occur when protein modifications are unknown. In this case, de novo sequencing can be performed, extracting data from the spectrum peaks derived from peptide fragment ions. There are two main types of proteomics strategies. Top-down proteomics — used when proteins of interest are separated from the rest of the sample before MS characterization. Bottom-up proteomics- also called “shotgun”- digests all proteins in a complex mixture, and all these peptides are analyzed. MS for proteomics has allowed comprehensive data to be generated on protein expression, protein interactions and defining sites of protein modifications. Quantitative measurements employing quantitative proteomics also measure dynamic changes or abundances of proteins or PTMs, which have been critical for studying mechanisms of diseases and the effects of treatment. The availability of public data depositories of MS-based proteomic data also consolidates experimental data from scientific articles.
Limitations of proteomics
Proteomics and MS have been crucial in experimentally validating most of the proteins predicted from the human genome. However, proteins still lack experimental validation, known as missing proteins (MPs). These MPs have been reported at the transcript level or by homology or prediction studies. These MPs may also be expressed at very low levels or in rare or tissue cell types(4). In addition, a relatively large amount of starting material is needed for proteomics analysis, and consequent bulk analysis may conceal crucial information about cellular subpopulations.
Spatial proteomics
"While proteomics approaches have facilitated the analysis of proteins present in cells and tissues, spatial proteomics have enabled the delineation of proteins’ spatial localization within cells, which has enhanced our understanding of their form and function." (5) Spatial proteomics allows the elucidation of the spatial distribution of molecules from tissues and can be used to target small tissue areas. In doing so, information can be generated on individual cells with distinct phenotypes. A spatial proteomic protocol amenable to most laboratories is proximity labelling (PL), which captures proteins from organelles or subcellular compartments. PL enzymes are engineered enzymes that, when expressed, can be used to distinguish protein-protein interactions as well as protein-nucleotide interactions and answer questions about protein localization. Two common PL enzymes are ascorbate peroxidase 2 (APEX2) and biotin ligases (BioID).
APEX is a peroxidase that can be fused to a protein of interest and target different subcellular locations. By using hydrogen peroxide (H2O2) as a catalyst, it can add biotin-phenol to endogenous proteins that are within 20nm of the APEX2 active site. This reaction is performed briefly to get a snapshot of the protein complexes from a subcellular location. BioID works in a similar way. The protein of interest is expressed as a fusion of BirA, which is a biotin ligase. BioID takes longer than APEX, taking 6-24 hours and labels proteins within 10nm of the enzyme. A streptavidin matrix can be used to capture and purify biotinylated proteins, followed by enzyme digestion to generate a bottom-up MS-based proteomics approach.
Mass spectrometry imaging (MSI)
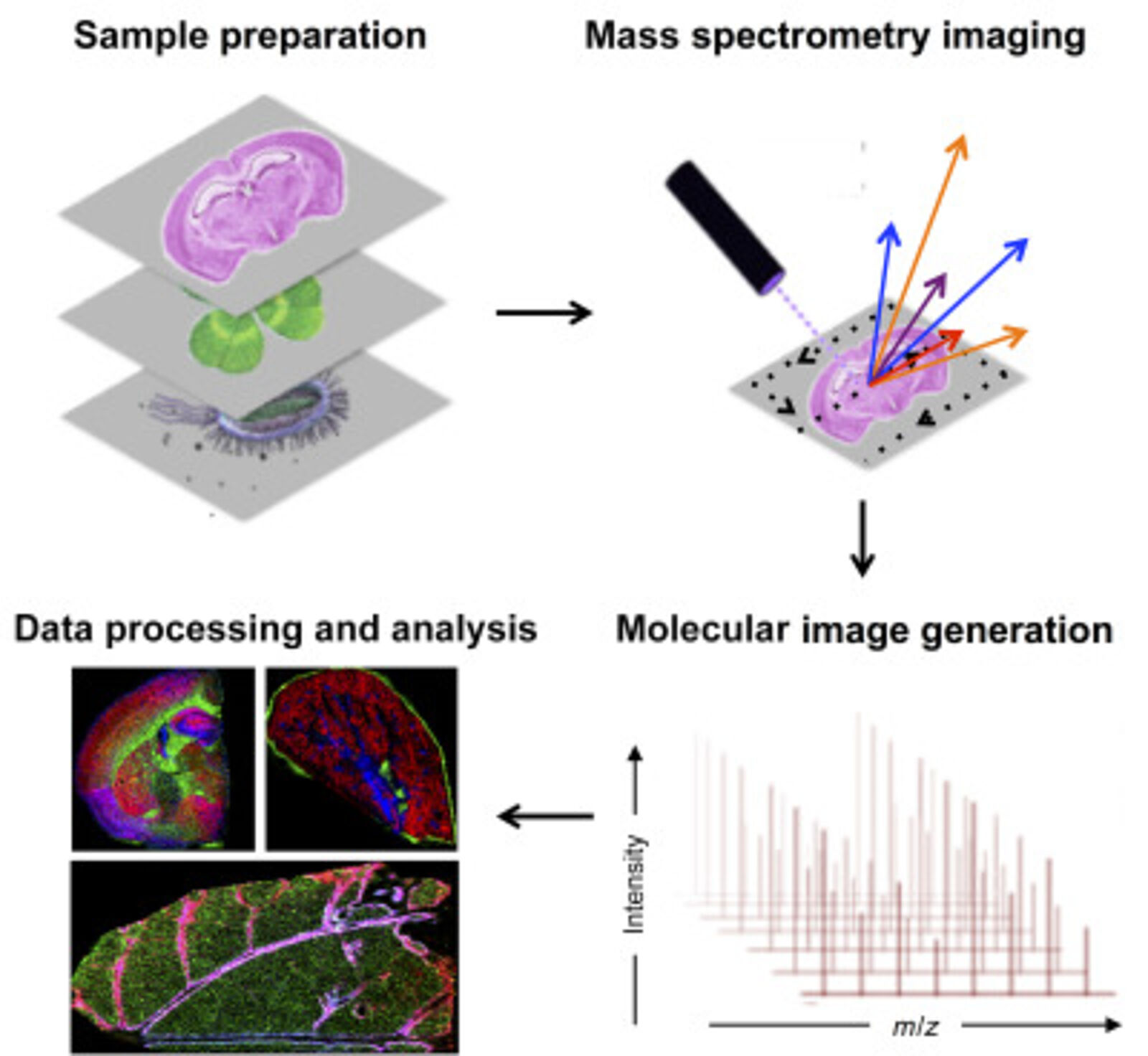
Mass spectrometry imaging (MSI) has become an important tool in spatial proteomics. MSI can map the spatial distribution of label-free analytes from a single cell in a specimen. A general MSI workflow consists of the following steps. Samples are first acquired and treated to maintain sample integrity. The sample can be frozen to reduce degradation and protein delocalization. Other preservative methods can also be used. Advances have been made to enable formalin-fixed paraffin-embedded (FFPE) samples to be more accessible for MSI. Embedding samples into a supporting medium may be used prior to sectioning to preserve tissue morphology, which is particularly important for fragile samples. Sample treatment includes adding digestion enzymes, matrix or derivatization agents, which depend on the protocol and instrument used. Once the grid on the surface of the sample is defined, a spectrum can be acquired. MS detection methods commonly used for MSI are MALDI and nanospray desorption electrospray ionization.
Spatial proteomics using MSI has been vital in studying differences in tissues. MSI has also been key in laser microdissection and has been combined with MSI to isolate small regions of tissues. This is particularly crucial in the clinical setting when analyzing tumours containing intra-tumor heterogeneity (6)
Antibodies can also be combined with MSI to target molecules of interest. For this, photocleavable mass tags are attached to antibodies and lectins. The tags are cleaved using UV light and ionized during MSI, which allows targeted imaging of selected markers with MALDI-MSI. This technique has coupled immunohistochemistry (IHC) with MALDI-MSI and has been shown to produce sensitive, highly multiplexed IHC analysis targeting a range of biomarkers in different tissues, including mouse brain, human tonsils and breast cancer (7). In addition, fluorophores can also be coupled to antibodies with mass tags allowing validation with immunofluorescence analysis.
Studying PTMs, such as phosphorylation, that are liable to spatial and temporal variation is challenging. Another method used for spatial proteomics is cell fractionation and purification combined with MS techniques. In a publication by Martinez-Val et al., subcellular fractionation combined with MS was used to study the spatiotemporal EGFR phospho-signaling dynamics in vitro in HeLa cells and in vivo in mouse tissues (8).This protocol preserves cellular phospho-networks and allows the high-throughput analysis of spatial dynamics.
In the next issue of the technical blog, we will discuss further examples of spatial proteomic workflows, the challenges faced in the field and how researchers are combating these challenges.
References
- Wilkins, M. R. et al. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev13, (1996).
- O’Farrell, P. H. High-resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry250, (1975).
- Shevchenko, A. et al. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two-dimensional gels. in Proceedings of the National Academy of Sciences of the United States of America vol. 93 (1996).
- Sjöstedt, E. et al. Integration of Transcriptomics and Antibody-Based Proteomics for Exploration of Proteins Expressed in Specialized Tissues. J Proteome Res17, (2018).
- www.nature.com/collections/daiceggbch
- Longuespée, R. et al. MALDI Imaging Combined with Laser Microdissection-Based Microproteomics for Protein Identification: Application to Intratumor Heterogeneity Studies. in Methods in Molecular Biology vol. 1788 (2018).
- Yagnik, G., Liu, Z., Rothschild, K. J. & Lim, M. J. Highly Multiplexed Immunohistochemical MALDI-MS Imaging of Biomarkers in Tissues. J Am Soc Mass Spectrom32, (2021).
- Martinez-Val, A. et al. Spatial-proteomics reveals phospho-signaling dynamics at subcellular resolution. Nat Commun12, (2021).
Figures
- O’Farrell, P. H. High-resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry250, (1975).
- Gregorich, Z. R.; Chang, Y. H.; Ge, Y. Pflugers Archiv-European Journal of Physiology 2014, 466, 1199-1209.
- De Raad, M., Northen,T. R., Bowen, B. P., Comprehensive Analytical Chemistry, Volume 82, (2018) doi.org/10.1016/bs.coac.2018.06.006
Suppliers
Absolute Antibody
Absolute Antibody is an expert in engineering recombinant antibodies for in vivo research. Their custom services include hybridoma sequencing, antibody engineering and expression.
Proteintech
Primary antibodies, nanobodies, cytokines & growth factors - all made in-house. Strict validation by western blot, ELISA and siRNA testing.
Bethyl Laboratories - Fortis Life Sciences applies B-cell sorting and recombinant DNA technology to deliver high quality recombinant rabbit monoclonal antibodies. The pillar strategy validation ensures that the antibody works in the stated applications.